

Downregulation of class I human leukocyte antigen (HLA)-restricted antigen presentation has been identified as mechanism of immune-escape in many malignant and non-malignant disorders. In idiopathic aplastic anemia (AA), evolution of immune-privileged paroxysmal nocturnal hemoglobinuria (PNH) clones has been attributed to immune escape due to deficiency of GPI-anchored protein in the context of T-cell mediated autoimmunity. However, other mechanisms of clonal selection may also operate with or independently of PNH. Our group first described the presence of both somatic uniparental disomy (UPD) and microdeletions of the HLA region leading to loss of heterozygozity (LOH) and/or haploinsuffciency.1 Later the proof-of-concept of somatic mutations in HLA class I was provided.2 Mechanistically, HLA LOH leads to loss of an allele involved in the presentation of immune-dominant peptides, while haploinsufficiency may decrease the presentation threshold. Moreover, the general level of individual structural diversity of HLA molecules may determine the ability to present diverse targets, eventually derived from auto-antigens, and functionally would operate in the opposite direction to HLA LOH.
In this scenario, we hypothesize that defects in both class I and II HLA loci may constitute different patterns of immune escape, reducing respectively CD8+ and CD4+ related activation and thus contributing to rescue hematopoietic stem cells from the immune attack. Furthermore, our idea is that the immune-escape environment may be related to the grade of HLA evolutionary divergence (HED), a metric that, accounting for the degree of structural diversity within a particular locus, represents an indirect measure of the antigenic landscape that the hematopoietic target cell is able to present (see abstract #:142693).
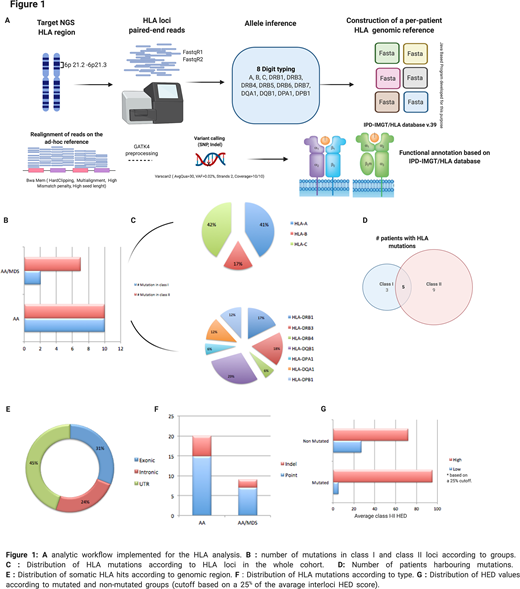
Using a deep targeted HLA NGS panel and a newly developed in-house bioinformatic pipeline (characterized by stringent criteria for alignment, preprocessing and variant calling in the HLA region, based on the IPD IMGT/HLA database, Fig.A), we studied a large cohort of patients with idiopathic bone marrow failures (AA n=75, AA/MDS=10). In addition, we determined the impact of inter-loci HED on the probability to acquire somatic hits in HLA genes.
Overall, 29 somatic HLA mutations were found in 16 patients (18%) at a median VAF of 11% (range: 2-93%):12 in class I (41%) and 17 in class II (59%), with 5 patients carrying mutations in both classes (Fig.B, C, D). The majority of those events (N=21, 72%) occurred in subjects also harbouring a PNH clone of small size (12 out 16 patients, median PNH clone size 1% [range:1-46%]). Most mutated loci were A and C for class I and DQB1 for class II (Fig. C, D); 9 mutations were identified as missense, with disruptive changes, 7 were intronic indels while 13 hits were localized in 5' or 3' untranslated regions (UTRs) (Fig.E, F). Through a computational prediction of the HLA regulatory domains involved in the UTR aberrations, we identified domains essential for the binding of GATA-1, RXRbeta, SP-1 and NFKB. The impairment of those regions may affect the transcription of HLA complexes. AA HLA mutant cases had more frequently a severe disease at diagnosis (severe AA: 81% vs. 60%, respectively in HLA mutated vs non mutated cases) and were in most part responders to immunosuppressive therapy (complete/partial responses: 75% vs 50% in HLA mutated vs non mutated patients). Within the AA/MDS group instead HLA mutations were found in 4 out of 10 patients (40%), including of note three -7/del7q cases.
Using Pierini and Lenz algorithm3 to determine inter-class HED, we found that HLA mutations tended to occur more often in patients with a high inter-class mean HED (94% vs 72% in non mutated group, p=.001, Fig. G), consistent with the idea that higher structural diversity of HLA molecules may induce more pervasive auto-immune responses, stronger immune pressure and ultimately the establishment of immune-escape mechanisms.
In summary, our results indicate the importance of class-I and -II HLA loci somatic hits as markers of autoimmunity and thereby the severity of the immune selection pressure, configuring possibly alternative mechanisms of immune-escape, in addition to immune privileged PNH clones. This environment may ultimately facilitate leukemic clonal expansion in AA-MDS setting.
Patel:Alexion: Other: educational speaker. Peffault De Latour:Apellis: Membership on an entity's Board of Directors or advisory committees; Alexion Pharmaceuticals Inc.: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Pfizer: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Novartis: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Amgen: Research Funding. Maciejewski:Novartis, Roche: Consultancy, Honoraria; Alexion, BMS: Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal